Usage
Overview
S2Omics is organized as a modular pipeline:
Histology Preprocessing (p1) – Scale and pad raw H&E stained images.
Superpixel Quality Control (p2) – Split image into superpixels and remove low-quality tiles.
Feature Extraction (p3) – Apply foundation model to obtain deep embeddings of image patches.
Histology Segmentation (p4) – Cluster embeddings into histology-based morphological clusters.
Cluster Merging (p5) – Merge highly similar clusters to reduce over-segmentation.
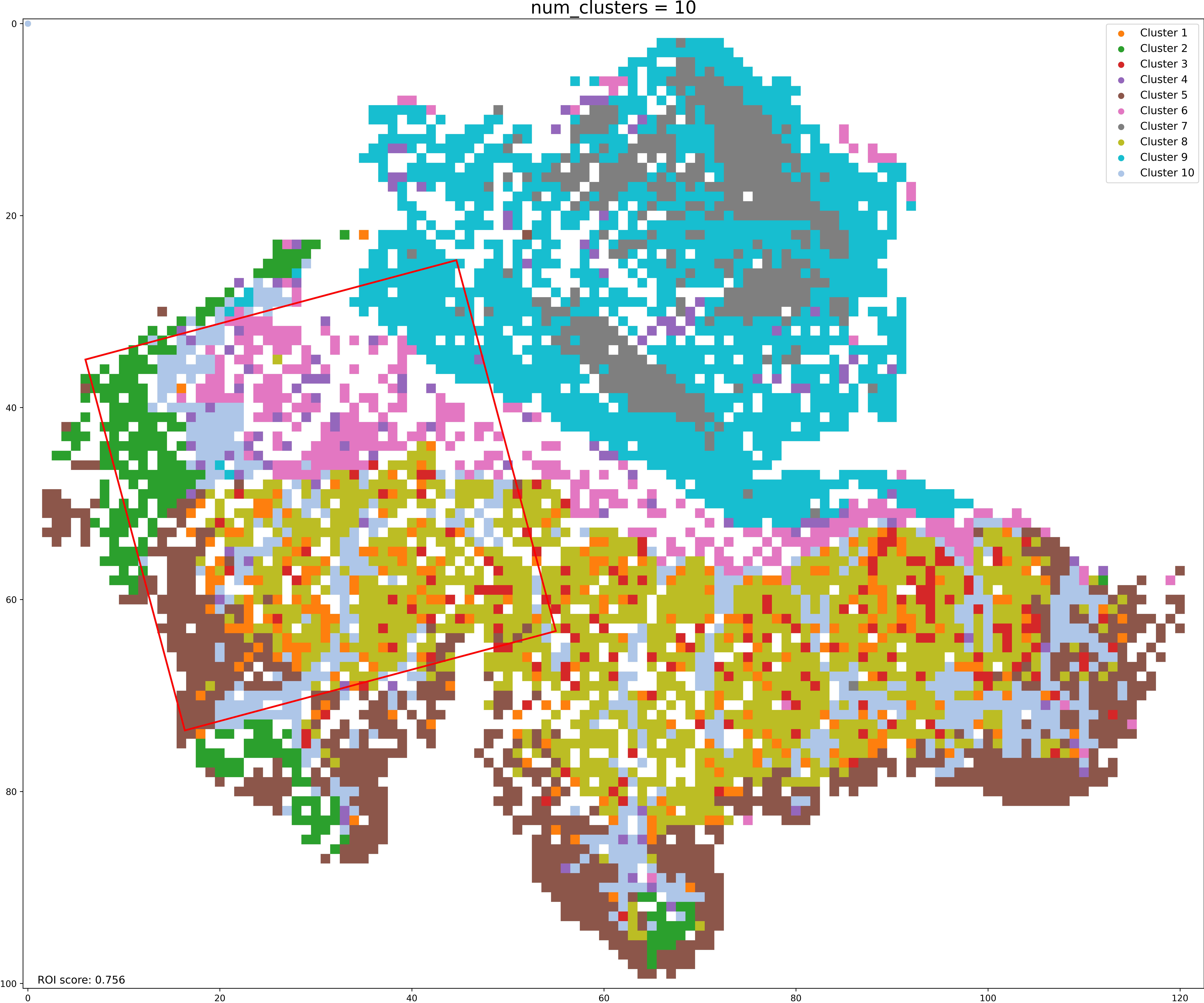
ROI Selection (p6) – Automatically select optimal Regions of Interest given constraints.
Label Broadcasting (p7) – Project cell type annotations from ROI-scale spatial omics data to the entire slide.
Installation
We recommend Python 3.11+.
git clone https://github.com/ddb-qiwang/S2Omics.git
cd S2Omics
conda create -n S2Omics python=3.11
conda activate S2Omics
pip install -r requirements.txt
# if GCC is very old:
pip install -r requirements_old_gcc.txt
Demo Data and Models
Download from Google Drive:
https://drive.google.com/drive/folders/1z1nk0sF_e25LKMyHxJVMtROFjuWet2G_?usp=sharing
Place both checkpoints and demo under S2Omics main folder.
Running the Full ROI Selection Pipeline
Use run_roi_selection.sh to execute all steps p1–p6.
Example (select 1 rectangular 6.5mm x 6.5mm ROI at downsampling step=10):
chmod +x run_*
./run_roi_selection_demo.sh
Typical output:
This runs:
p1_histology_preprocess.py - Input: Folder with he-raw.jpg, pixel-size-raw.txt - Output: he-scaled.jpg, padded he.jpg.
p2_superpixel_quality_control.py - Splits he.jpg into patch_size superpixels (default 16×16 px). - Applies density filtering and texture analysis to remove low-quality tiles.
p3_feature_extraction.py - Loads foundation model (UNI/Virchow/GigaPath). - Extracts two-level embeddings (global 224×224, local patch-level).
p4_get_histology_segmentation.py - Clusters PCA-reduced embeddings into morphological clusters. - Methods: kmeans, fuzzy c-means, Louvain, Leiden, etc.
p5_merge_over_clusters.py - Merges clusters with high similarity (hierarchical-linkage based) to target cluster count.
p6_roi_selection_rectangle.py / p6_roi_selection_circle.py - Uses search + scoring system (scale, coverage, balance) to select ROIs automatically or for given num_roi.
Running Cell Label Broadcasting
Prerequisite: you have spatial omics ROI-level annotations (annotation_file.csv).
Example:
./run_label_broadcasting_demo.sh
Output example:

This runs p1-p7:
p7_cell_label_broadcasting.py – Loads histology features from ROI-scale omics and from whole-slide image, trains a small autoencoder-based classifier, and predicts cell type for every valid superpixel across the slide.
Input File Formats
he-raw.jpg – raw histology image.
pixel-size-raw.txt – microns/pixel for raw image.
pixel-size.txt – target microns/pixel after rescaling.
annotation_file.csv (optional) – Required for label broadcasting. Columns: super_pixel_x, super_pixel_y, annotation.
Example annotation file: