Tutorial 1: Design VisiumHD experiment for a colorectal cancer section
Please download according demo data from following link and place it under the demo folder:
google drive: https://drive.google.com/drive/folders/1z1nk0sF_e25LKMyHxJVMtROFjuWet2G_?usp=drive_link
Please also download the checkpoint file for the pathology foundation model and place it under the checkpoints folder
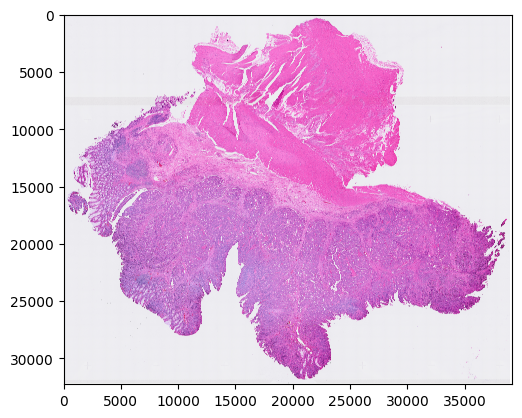
Step 1: Preprocess the H&E image
Make sure the physical size of each pixel is 0.5 micron
[1]:
import sys
sys.path.append('..')
from s2omics.p1_histology_preprocess import histology_preprocess
prefix = '../demo/Tutorial_1_VisiumHD_ROI_selection_colon/'
histology_preprocess(prefix, show_image=True)
Image loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/he-raw.jpg
Rescaling image (scale: 1.000)...
244 sec
../demo/Tutorial_1_VisiumHD_ROI_selection_colon/he-scaled.jpg
../demo/Tutorial_1_VisiumHD_ROI_selection_colon/he.jpg
Preprocessed H&E image saved!
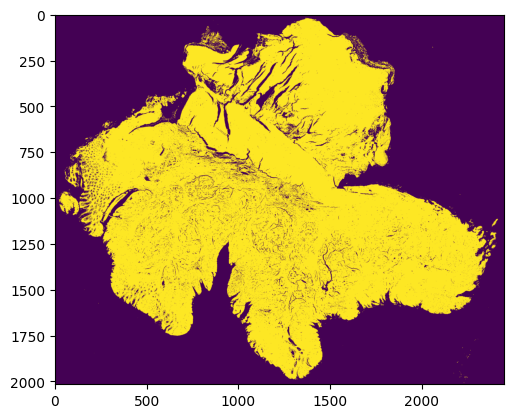
Step 2: Quality control for all superpixels
Superpixels are 8 microns * 8 microns square-shaped pseudo cells
We use our new QC package HistoSweep for this procedure
[2]:
from s2omics.p2_superpixel_quality_control import superpixel_quality_control
save_folder = '../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output'
superpixel_quality_control(prefix, save_folder, show_image=True)
Image loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/he.jpg
0 0
../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/shapes.pickle
[compute_metrics_memory_optimized] Current memory: 0.1839 GB; Peak memory: 3.7391 GB
[compute_low_density_mask] Current memory: 0.0046 GB; Peak memory: 0.2989 GB
Total selected for density filtering: 415494
✅ Entropy map saved as 'glcm_entropy_map_colored.png'
✅ Energy map saved as 'glcm_energy_map_colored.png'
✅ Homogeneity map saved as 'glcm_homogeneity_map_colored.png'
=== GLCM Metric Means ===
homogeneity energy entropy
0 0.219866 0.030197 0.891417
1 0.482113 0.149341 0.676108
2 0.366501 0.076353 0.775128
3 0.584400 0.309048 0.559195
=== Cluster Scores ===
Cluster 0: Score = -0.6414
Cluster 1: Score = -0.0447
Cluster 2: Score = -0.3323
Cluster 3: Score = 0.3343
=== Number of Observations per Cluster ===
Cluster 0: 127419
Cluster 1: 68422
Cluster 2: 106474
Cluster 3: 31172
Total: 333487
✅ Clustered texture map saved as 'cluster_labels_colored.png'
[run_texture_analysis] Current memory: 0.0049 GB; Peak memory: 11.7710 GB
[run_ratio_filtering] Current memory: 0.0046 GB; Peak memory: 0.1103 GB
(4935168,)
✅ Final masks saved in: HistoSweep_output
[generate_final_mask] Current memory: 0.0000 GB; Peak memory: 1.1907 GB
Running successfully!
../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/qc_preserve_indicator.pickle
Step 3: Histology feature extraction
[3]:
from s2omics.p3_feature_extraction import histology_feature_extraction
# down_samp_step: the down-sampling step,
# default = 10 refers to only extract features for superpixels whose row_index and col_index can both be divided by 10 (roughly 1:100 down-sampling rate).
# down_samp_step = 1 means extract features for every superpixel
histology_feature_extraction(prefix, save_folder,
foundation_model='uni',
ckpt_path='../checkpoints/uni/',
device='cuda:0',
batch_size=32,
down_samp_step=10,
num_workers=4)
/data1/msyuan/anaconda3/envs/S2Omics/lib/python3.11/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
Histology foundation model loaded!
Foundation model name: uni
Start extracting histology feature embeddings...
Image loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/he.jpg
../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/num_patches.pickle
0%| | 0/1547 [00:00<?, ?it/s]
Batch 0:
Shape of patches: torch.Size([32, 3, 224, 224])
Shape of positions[0]: torch.Size([32])
Content of positions[0][:10]: tensor([0, 0, 0, 0, 0, 0, 0, 0, 0, 0])
Content of positions[1][:10]: tensor([ 0, 160, 320, 480, 640, 800, 960, 1120, 1280, 1440])
Shape of feature_emb: torch.Size([32, 197, 1024])
Shape of patch_emb: torch.Size([32, 1024, 14, 14])
100%|█████████▉| 1546/1547 [20:58<00:00, 1.29it/s]
Part 0 patch number: 49490
100%|██████████| 1547/1547 [21:02<00:00, 1.23it/s]
../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/uni_embeddings_downsamp_10_part_0.pickle
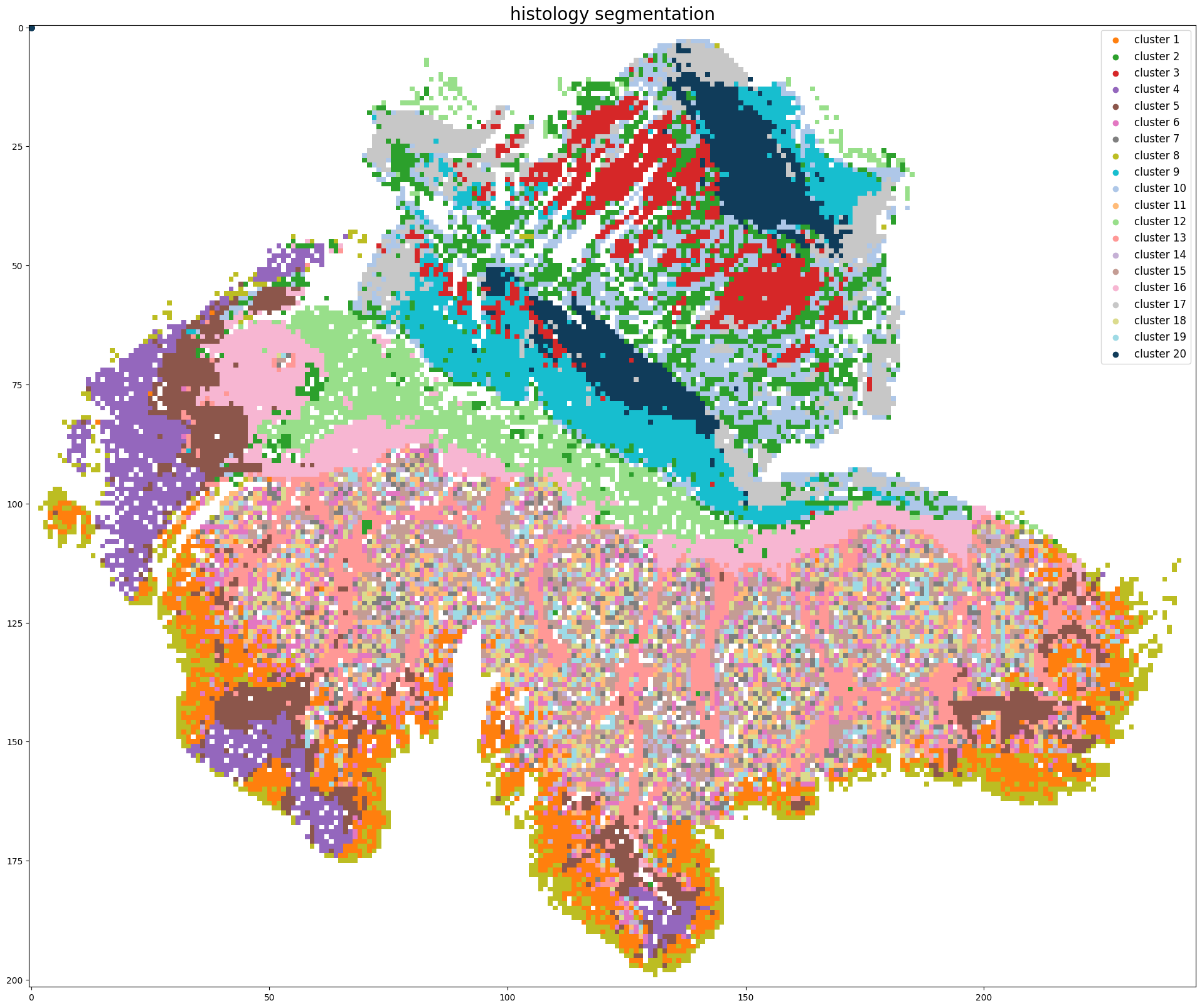
Step 4: Histology segmentation
[4]:
from s2omics.single_section.p4_get_histology_segmentation import get_histology_segmentation
get_histology_segmentation(prefix, save_folder,
foundation_model='uni',
down_samp_step=10,
clustering_method='kmeans',
n_clusters=20)
Pickle loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/shapes.pickle
Pickle loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/qc_preserve_indicator.pickle
Loading histology feature embeddings...
Pickle loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/uni_embeddings_downsamp_10_part_0.pickle
Sucessfully loaded and normalized all histology feature embeddings!
Start segmenting the histology image, clustering method: kmeans
../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/cluster_image.pickle
../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/linkage_matrix.pickle
Segmentation image is stored at: ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/image_files/cluster_image_num_clusters_20.jpg
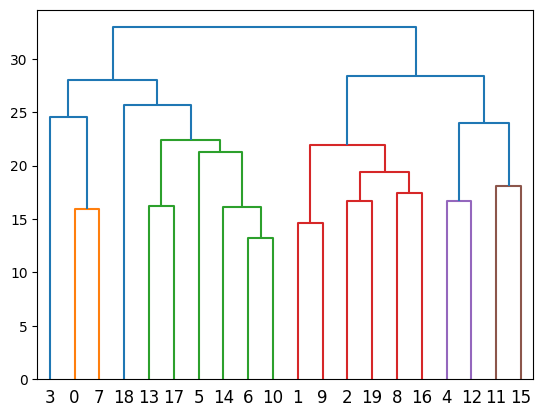
Step 5: Merge over-clusters
[5]:
from s2omics.single_section.p5_merge_over_clusters import merge_over_clusters
merge_over_clusters(prefix, save_folder,
target_n_clusters=15)
Pickle loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/shapes.pickle
Pickle loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/qc_preserve_indicator.pickle
Pickle loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/cluster_image.pickle
Merging over-clusters...
Pickle loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/linkage_matrix.pickle
../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/adjusted_cluster_image.pickle
Combined the original 20 clusters into 15 clusters.
Adjusted segmentation image is stored at: ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/image_files/adjusted_cluster_image_num_clusters_15.jpg
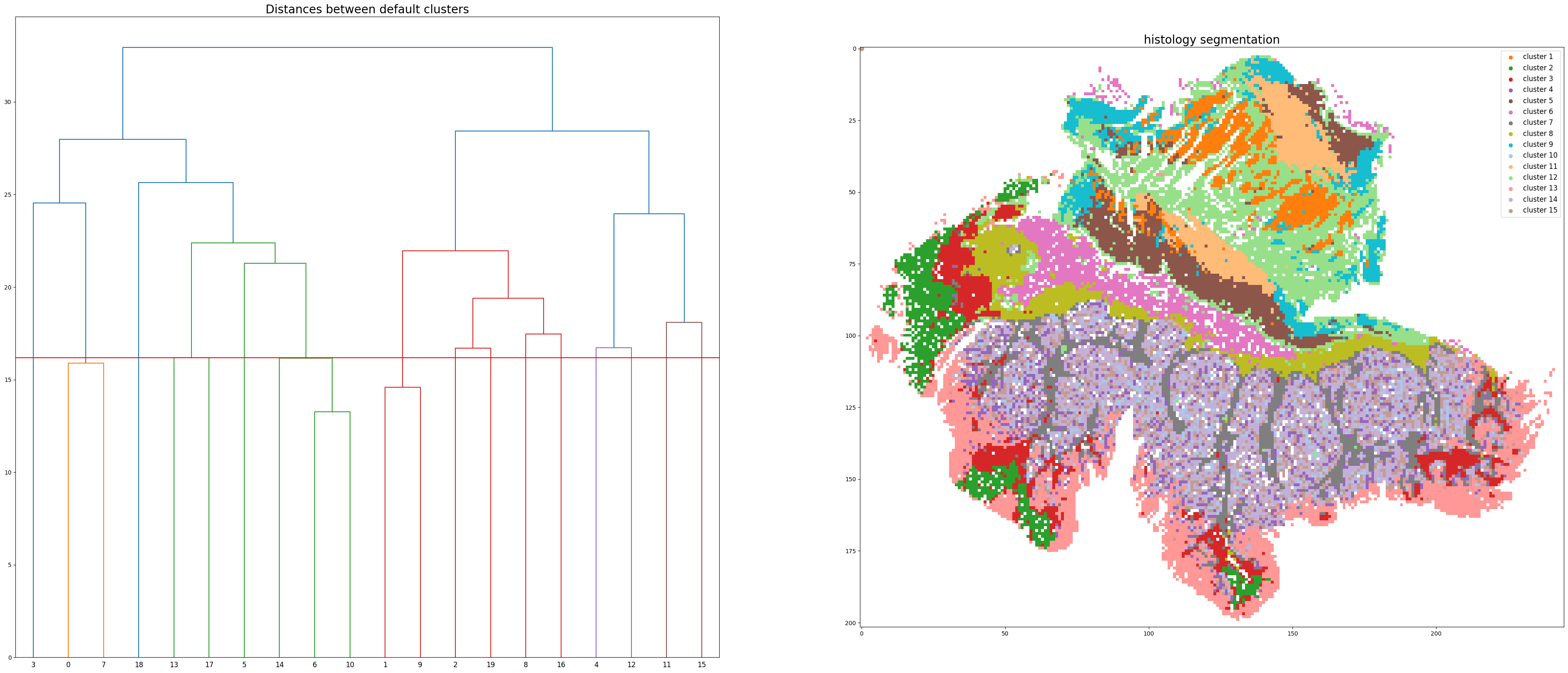
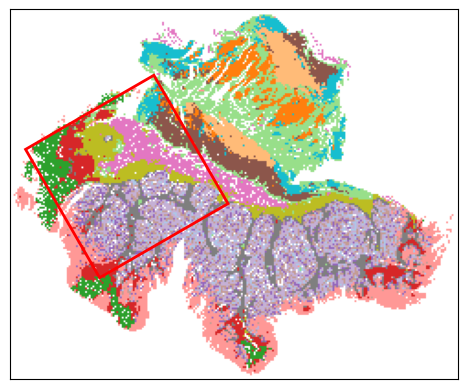
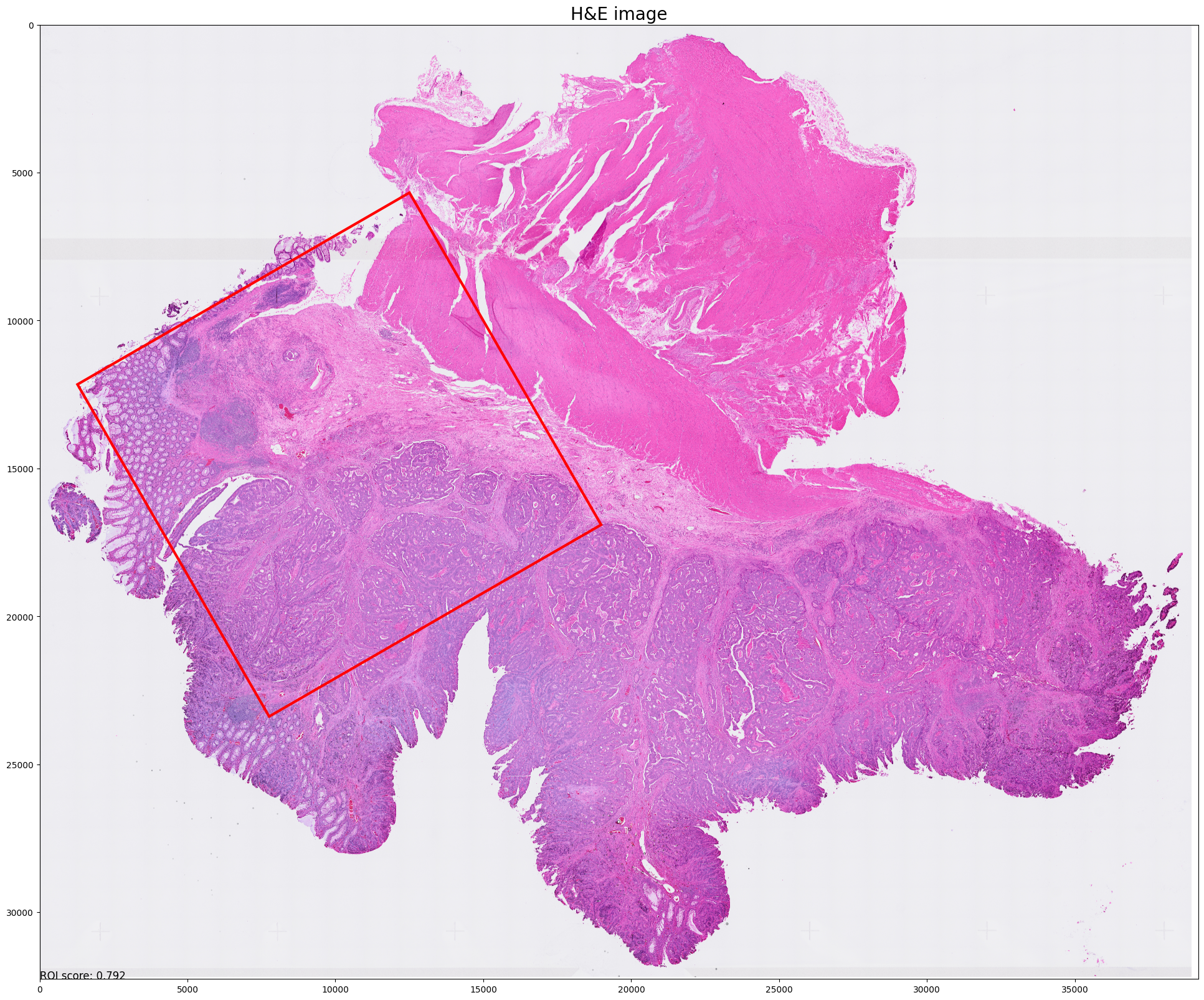
Step 6: Select best ROI for VisiumHD experiment
[6]:
from s2omics.single_section.p6_roi_selection_rectangle import roi_selection_for_single_section
# fusion_weights: the weight of three scores, default=[0.33,0.33,0.33], the sum of three weights should be equal to 1 (if not they will be normalized)
# positive_prior, negative_prior: prior information about interested and not-interested histology clusters, default = [],[]
# prior_preference: the larger this parameter is, S2Omics will focus more on those interested histology clusters, default= 1
roi_selection_for_single_section(prefix, save_folder,
down_samp_step=10,
roi_size=[6.5,6.5],
rotation_seg=6,
num_roi=1, #0 refers to automatiacally determine the number of ROI
fusion_weights=[0.33,0.33,0.33],
emphasize_clusters=[], discard_clusters=[],
prior_preference=1)
Image loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/he.jpg
Pickle loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/shapes.pickle
Pickle loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/qc_preserve_indicator.pickle
Pickle loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/adjusted_cluster_image.pickle
Sampling ROI candidates...
100%|██████████| 800/800 [00:00<00:00, 2969.77it/s]
Current best ROI: [[[76, 8], [146, 49], [105, 119], [35, 78]]]
roi score: 0.7923578790248927
scale score: 0.6245726221007392
valid score: 0.9455532908082276
balance score: 0.8423550596065301
Current number of ROIs is 1.
Find the best 1 ROI(s) with:
ROI score: 0.7923578790248927
Scale score: 0.6245726221007392
Coverage score: 0.9455532908082276
Balance score: 0.8423550596065301
../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/roi_selection_detailed_output/rectangle_roi_size_6.5_6.5/prior_preference_1/best_roi.pickle
Best ROI on histology segmentation image is stored at ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/main_output/best_roi_on_histology_segmentations.jpg
Best ROI on H&E image is stored at ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/main_output/best_roi_on_he.jpg
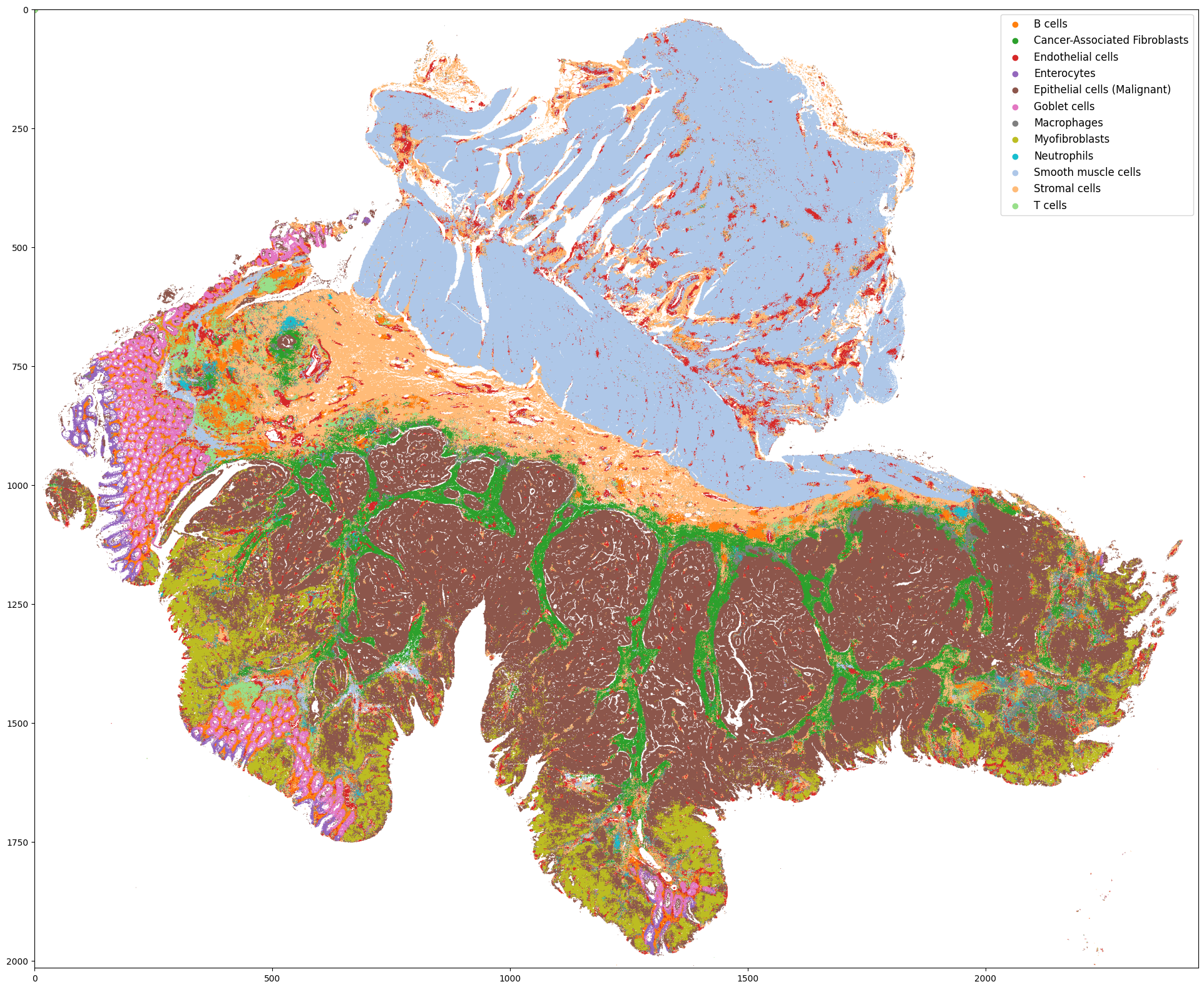
(Optional) Step 7: Cell type label broadcasting
** This step is based on the cell type label obtained from spatial omics experiment of selected ROI
** In this case (VisiumHD experiment design), the experiment can be conducted directly on the H&E slide, so we only need to broadcast the label from the ROI to the rest part of the slide
** But in many other situations, researchers may need to conduct experiment on another, adjacent slide, so we have to broadcast the label within the ROI from one slide to another slide.
Notice: this step may cost a lot of time as we need to extract the histology features for each small superpixel
[ ]:
# Run this cell if feature extraction isn't run with down_samp_step=1
import sys
sys.path.append('..')
from s2omics.p3_feature_extraction import histology_feature_extraction
# down_samp_step: the down-sampling step,
# default = 10 refers to only extract features for superpixels whose row_index and col_index can both be divided by 10 (roughly 1:100 down-sampling rate).
# down_samp_step = 1 means extract features for every superpixel
histology_feature_extraction(prefix, save_folder,
foundation_model='uni',
ckpt_path='../checkpoints/uni/',
device='cuda:0',
batch_size=32,
down_samp_step=1,
num_workers=4)
[7]:
from s2omics.single_section.p7_cell_label_broadcasting import label_broadcasting
# SO_datapath is the path to spatial omics data folder which should contain preprocessed H&E image and annotation_file.csv
## if the H&E image is not preprocessed, please refer to step 1 and step 2
# WSI_datapath is the path to original whole slide H&E image
# this step is to transfer the cell type label inside the ROI of spatial omics data to the whole slide H&E image
label_broadcasting(WSI_datapath=prefix, WSI_save_folder=save_folder,
SO_datapath=prefix, SO_save_folder=save_folder,
WSI_cache_path='', SO_cache_path='',
foundation_model='uni', device='cuda:0')
Pickle loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/shapes.pickle
Pickle loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/qc_preserve_indicator.pickle
Loading histology feature embeddings of the Spatial Omics data...
Pickle loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/uni_embeddings_downsamp_1_part_0.pickle
Sucessfully loaded all histology feature embeddings of the Spatial Omics data!
Pickle loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/shapes.pickle
Pickle loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/qc_preserve_indicator.pickle
Loading histology feature embeddings of the whole-lide H&E data...
Pickle loaded from ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_output/pickle_files/uni_embeddings_downsamp_1_part_0.pickle
Sucessfully loaded all histology feature embeddings of the whole-lide H&E data!
Start training the label transferring model...
Epoch [20] loss: 0.609, train accuracy 0.754
Epoch [40] loss: 0.476, train accuracy 0.808
Epoch [60] loss: 0.504, train accuracy 0.836
Epoch [80] loss: 0.455, train accuracy 0.846
Epoch [100] loss: 0.442, train accuracy 0.861
Finished Training
Predicted cell type distribution for the whole-slide H&E data is stored at: ../demo/Tutorial_1_VisiumHD_ROI_selection_colon/S2Omics_whole_slide_prediction.jpg
[ ]: